服务热线:
13600366215
13600366215






王老师
13600366215
gary.wang@bonnier.net.cn
微信:Bonnier-Gary
www.bonnier.net.cn
中国广东省江门市江海区海伦心苑1-3203

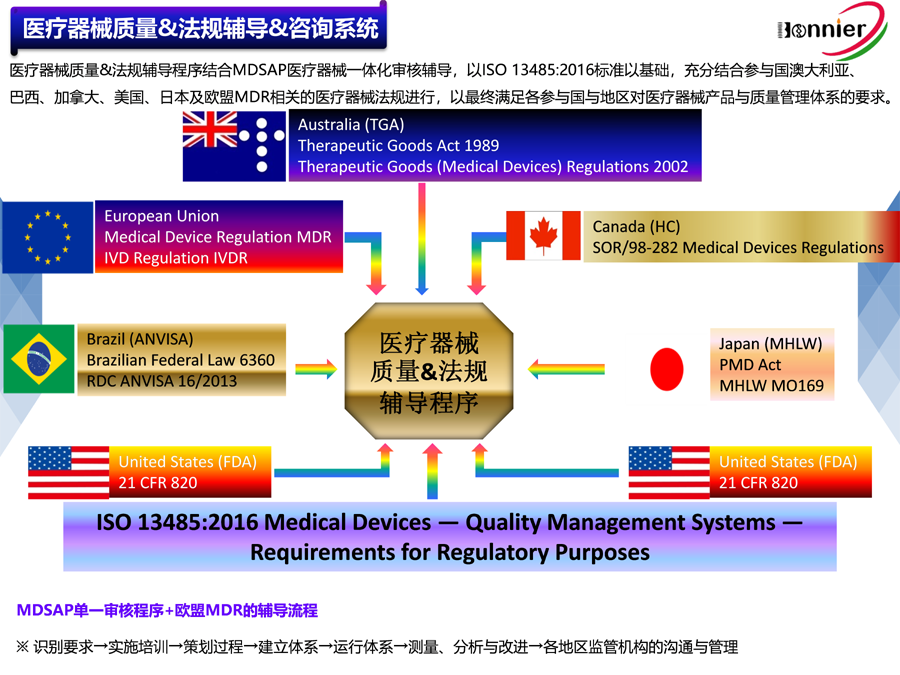
澳大利亚医疗器械的注册由治疗用品管理局(Therapeutic Goods Administration,简称TGA)负责管理,旨在确保医疗器械的安全性和有效性。以下是详细的注册流程:
1.确定产品分类:根据2002年《治疗商品(医疗器械)条例》,医疗器械被分为I类、IIa类、IIb类、III类和IV类。不同类别的医疗器械需要满足不同的注册要求和审核流程。
2.指定澳大利亚授权代表(Sponsor) :非澳大利亚本地公司必须指定一个澳大利亚授权代表来完成注册工作。授权代表负责与TGA沟通、提交注册申请,并处理后续的监管事务。
3.准备技术文件:制造商需准备详细的技术文件,包括产品描述、设计验证、临床评估报告、风险管理文件等,这些文件需符合TGA的技术标准和法规要求。
4.建立质量管理体系:制造商需确保拥有ISO13485:2016认证或MDSAP认证。
5.提交注册申请:通过TGA的电子系统(如TBS系统)提交注册申请,包括技术文件、澳大利亚合格声明等。
6.技术评估与审查:TGA将对提交的注册申请进行技术评估和审查,可能涉及书面审查、技术数据评估,高风险设备可能有现场检查。
7.生物相容性评估:对于与人体接触的医疗器械,TGA会要求进行生物相容性评估,以确保其不会对人体造成不良反应或毒性。
8.获得注册证书:如果医疗器械符合TGA的要求,TGA将颁发相应的注册证书或许可证,允许其在澳大利亚市场上销售和使用。
9.持续监管与合规:获得注册后,制造商需要确保持续遵守澳大利亚的相关法规和标准,接受TGA的监管和检查,包括定期更新技术文件、报告产品变更、回应TGA的要求等。
需要注意的是,澳大利亚的医疗器械注册流程可能较为复杂和耗时,通常需要几个月的时间完成审批。此外,不同类别的医疗器械所需的时间和费用也有所不同,例如I类和IIa类通常需要约4周,IIb类需要6周,III类和有源植入式医疗器械大约需要6个月。
要满足美国FDA的QSR 820(即21 CFRPart 820)要求,医疗器械制造商需要遵循一系列严格的质量管理体系规范。以下是详细的步骤和建议:
1.理解QSR 820的要求(21 CFR Part 820—QUALITY MANAGEMENT SYSTEMREGULATION)
QSR 820是美国食品药品监督管理局(FDA)制定的医疗器械质量体系法规,旨在确保医疗器械从设计、制造到服务的整个生命周期中保持安全和有效。该法规包括多个子系统,如管理控制、纠正和预防措施(CAPA)、设计控制、生产和过程控制、设备设施控制、材料控制以及文件记录和变更控制等。
2.建立符合QSR 820的质量管理体系(21 CFR Part 820—QUALITY MANAGEMENT SYSTEMREGULATION)
制造商需根据自身业务范围,建立并实施一个全面的质量管理体系,涵盖所有适用的条款。例如,对于Ⅰ类器械,设计控制仅需遵守特定的要求。此外,制造商应确保其体系能够覆盖产品的设计、制造、包装、标签、储存、安装和服务等环节。
3. 21 CFR Part 820—QUALITYMANAGEMENT SYSTEM REGULATION与ISO 13485标准的协调
QSR 820与ISO 13485:2016标准在实质要求上相似,但QSR 820将风险管理要求集成到质量管理的各个方面。因此,制造商可以参考ISO 13485标准来建立或优化其质量管理体系,以更好地符合QSR 820的要求。
4.文件管理与记录控制
文件管理是QSR 820的核心要求之一。制造商需确保所有与产品相关的文件都经过授权人员的审查和批准,并及时更新过时的文档。此外,所有操作程序和记录都必须保存以备FDA审核。
5.纠正和预防措施(CAPA)
制造商需建立并维护CAPA程序,包括调查、分析、确定纠正和预防措施、验证执行效果以及记录所有相关活动。这有助于持续改进产品质量和安全性。
6.设备和过程控制
对于生产设备和制造过程,制造商需确保所有设备符合规定,并定期进行校准和维护。同时,生产过程需严格按照既定程序执行,以避免任何可能导致产品不合格的因素。
7.培训与内部审核
制造商需对员工进行QSR 820相关的培训,确保他们理解并能够执行相关要求。此外,定期进行内部审核和管理评审,以发现并解决潜在问题。
8.应对FDA审核与检查
在FDA进行工厂检查时,制造商需准备好迎接审核,包括提供完整的文件记录、模拟审核以及陪同FDA检查官进行现场检查。对于检查中发现的不符合项,需及时整改并提交回复报告。
9.遵守其他相关法规
除了QSR 820(21 CFR Part 820—QUALITY MANAGEMENT SYSTEM REGULATION),制造商还需满足其他相关法规的要求,如FDA的510(k)注册程序、UDI(唯一设备标识)等。
通过以上步骤,制造商可以逐步建立和完善符合QSR 820要求的质量管理体系,从而确保其产品在美国市场的安全性和有效性,并顺利通过FDA的审核与检查。
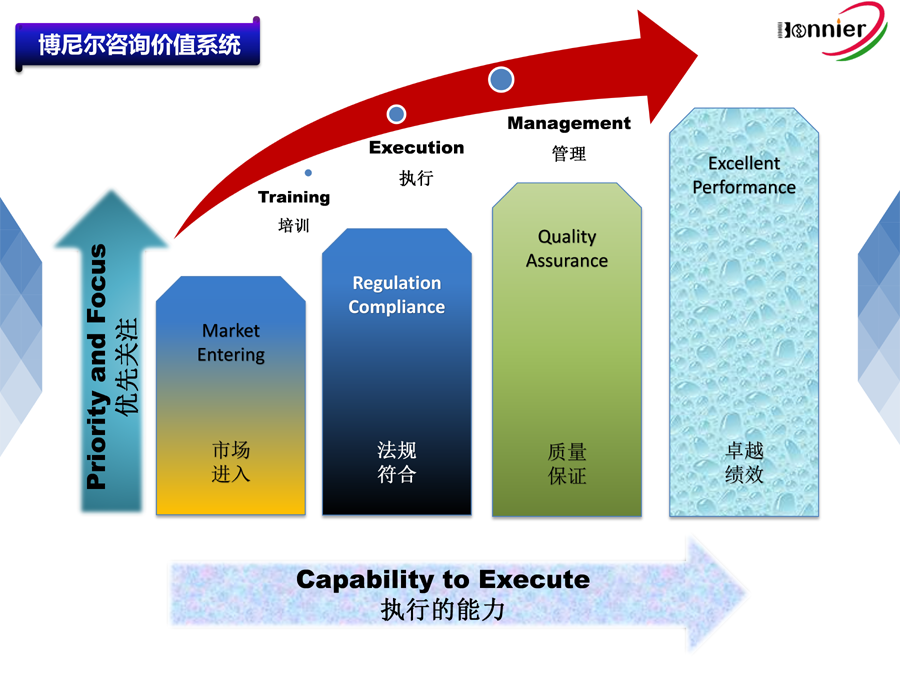
博尼尔质量管理咨询(江门)中心擅长的业务范围:
※医疗器械注册咨询辅导服务
医疗器械美国FDA 510k注册咨询辅导;
医疗器械加拿大HC MDL注册咨询;
医疗器械澳大利亚TGA注册咨询;
医疗器械欧盟MDR/IVDR CE注册咨询。
※医疗器械质量&法规管理体系咨询辅导服务:
ISO13485医疗器械质量管理体系咨询辅导;
医疗器械欧盟MDR/IVDR咨询辅导;
医疗器械MDSAP认证咨询;
医疗器械美国QSR820咨询辅导。
※医疗器械授权代表服务:
英国授权代表UKRP;
瑞士授权代表CHREP;
美国代理人FDA Agent;
欧盟授权代表ECREP。