服务热线:
13600366215
13600366215






王老师
13600366215
gary.wang@bonnier.net.cn
Wechat: Cognitive-Level
www.bonnier.net.cn
中国广东省江门市江海区海伦心苑1-3203

Medical Device Delegate(Medical Device Authorised Representative in China)
Medical Device entering the Chinese market offers tremendous opportunity for medical device companies, but it is not without challenges. Having a partner with experience and expertise in the Chinese marketplace is essential. It can be the difference between success and failure. Our experienced consultants can provide insight and guide you through the National Medical Products Administration (NMPA‘s old name is CFDA) in China.
The role of Medical Device Authorised Representative in China
Ⅰ Pre-market of medical device: To manage the registration process with NMPA on behalf of medical device manufacturer, to be the conjunction between NMPA and manufacturer in communications with the foreign establishment and product registration.
Ⅱ Post-market of medical device: To communicate with NMPA or related parties on behalf of medical device manufacturer,responding to questions concerning the foreign establishment´s devices that are imported into China market, to assist the investigation to medical device adverse event.
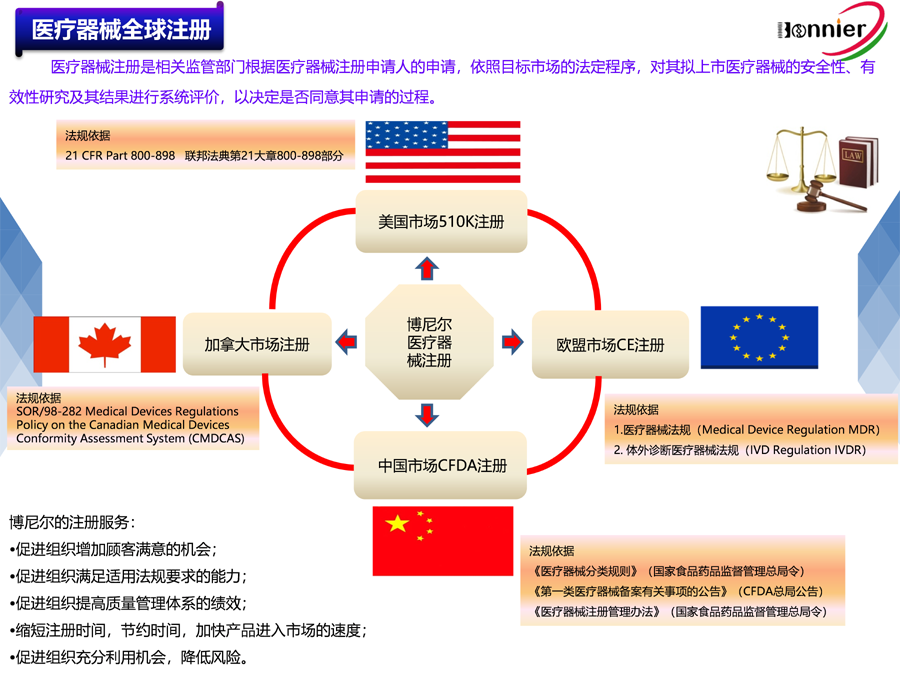
Medical Device and IVD Registration in China
Medical devices are regulated by the NMPA,which is under State Administration for Market Regulation. Manufacturers must register their devices with the NMPA before selling or distributing in China. The NMPA reviews all device applications and has strict requirements for submission documentation, testing and clinical data.
Your device classification determines the documentation required for your NMPA submission. Classification is based on CFDA regulation and the NMPA classification database. Device classification and their registration requirements are as follows:
Class I –registration dossier – self testing reports
Class II –full registration dossier and technical review
Class III– full registration dossier and technical review and clinical data
Manufacturers must submit a technical dossier that includes detailed technical information, clinical data and quality documentation. China requires in-country testing for all Class II and III devices, although the NMPA may accept some of your existing testing reports. Testing requirements vary depending on your device type.
We can Identify NMPA Testing and Clinical data Requirements in China
Bonnier is fully equipped to help youcomply with Chinese testing and clinical conformity requirements for your device before you start the registration process. Here´s how we can help:
1.Provide initial assessment to determine registration requirements and suggested clinical conformity route
2.Assist with identifying qualified testing centers in China and establishing the product technical requirement
3.Perform clinical literature review and prepare your CER, if needed
4.Advise you regarding clinical data requirements if you must perform clinical trials in China
5.Prepare registration submission documentation and submit to NMPA
6.Medical Device delegate in China to keep the compliance with NMPA regulation consistently
China Medical Device Regulatory Strategy Report
China´s medical device market is regulated by the NMPA, which is under State Administration for Market Regulation. All medical devices must receive CFDA approval before they can be marketed in the country, though Class I devices aresubject to a simpler notification process.
Let Bonnier Assist You in Navigating Medical Device Registration in China and the NMPA Approval as Medical Device Agent.
Bonnier relies on our in-house consultants as well as industry and regulatory contacts, to provide your Regulatory Pathway service. We offer incisive, actionable analysis including:
1.NMPA Regulatory Background
2.Product Assessment Based on NMPA criteria
3.Device Classification
4.Medical Device Registration Requirements
5.Labeling and Language Requirements
6.Costs and Timeframes
7.Post-Market Vigilance Requirements
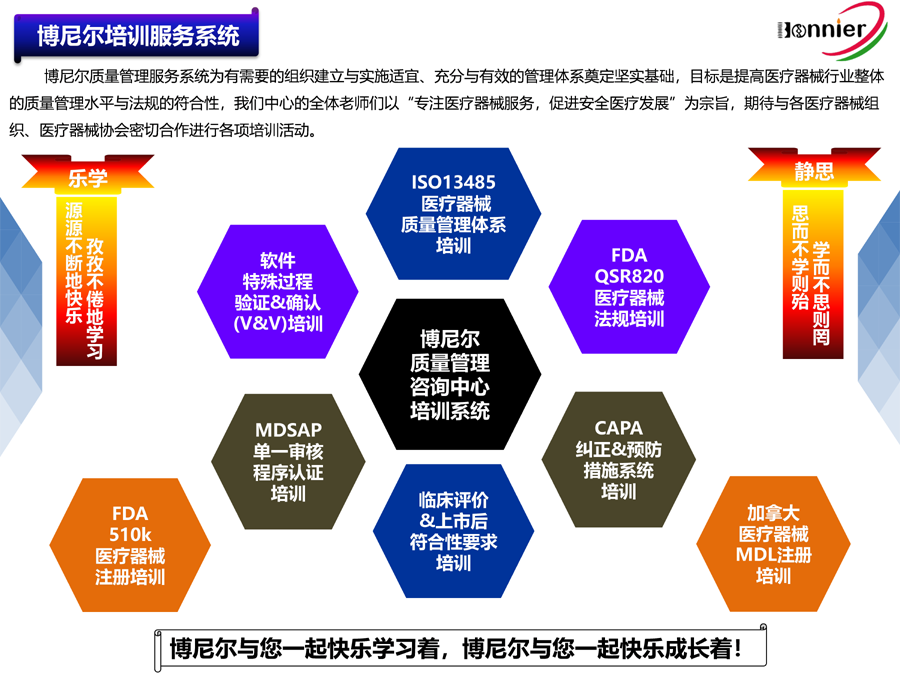
Main Business for Bonnier:
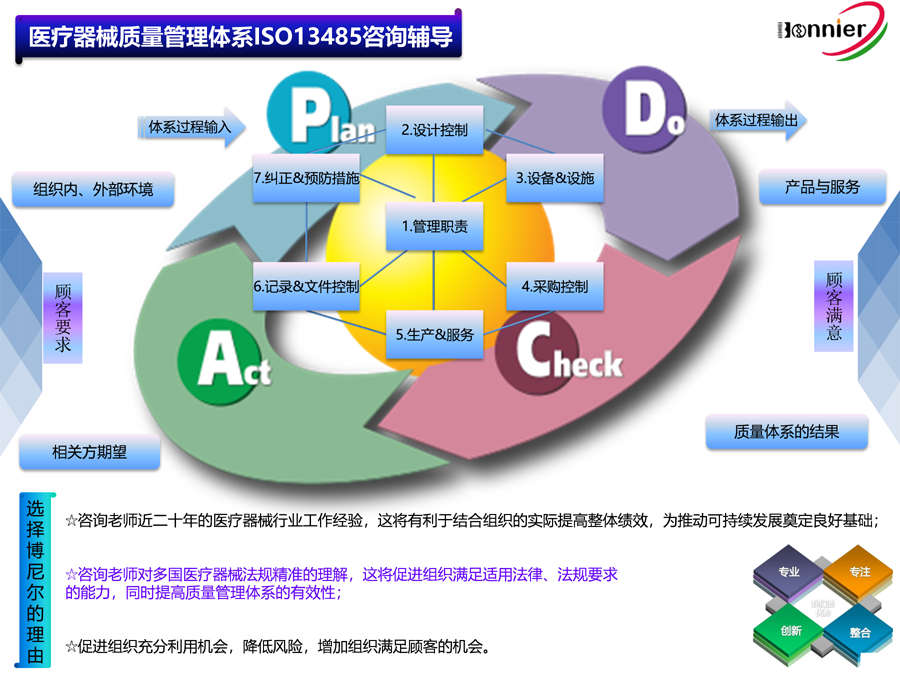
1.Consultant Service for ISO13485 Quality Management System
2.Consultant Service for Medical Device Single Audit Program (MDSAP)
3.Consultant Service for the Accreditation of FDA CFR820 Regulation Requirement and Accompanying Service of Factory Auditing
4.Overworld Medical Device Registration Service, Including USA 510k, Europe CE, Canada MDLand China NMPA Registration
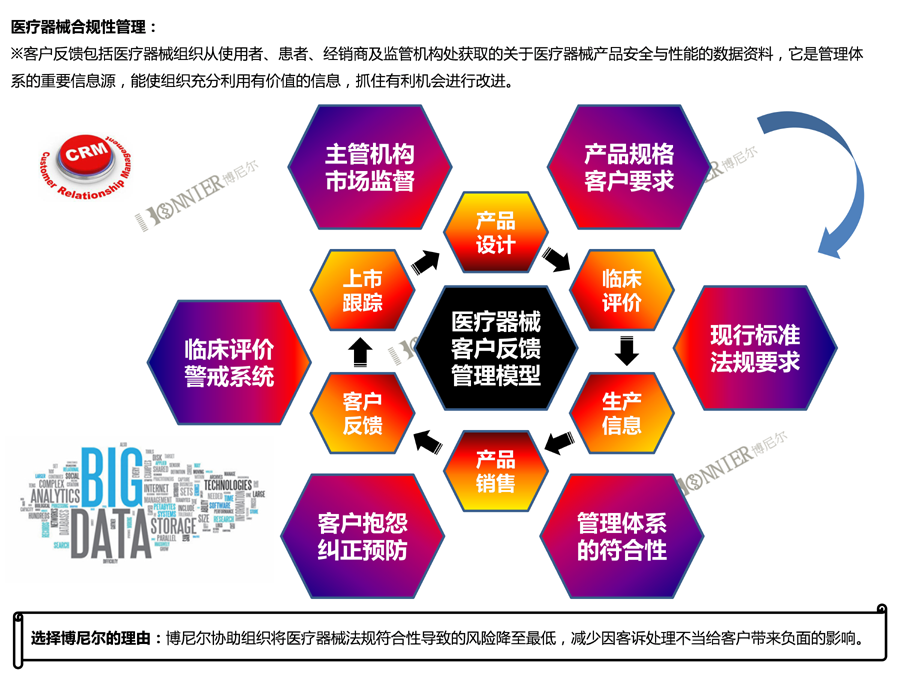
5.Medical Device Post Market Service, as Medical Device Adverse Event Reporting, Post-market Cliniclal Follow-up Service etc
6.Training for Medical Device Regulation and Quality Tools, as MDSAP regulation, EU MDR, CAPA System, Verification & Validation, Risk Management etc.
7.The Medical Device Agent in China will assist overall registration in China for classⅠⅡⅢ Medical Device and on regulation compliance in China