服务热线:
13600366215
13600366215






王老师
13600366215
gary.wang@bonnier.net.cn
Wechat: Cognitive-Level
www.bonnier.net.cn
中国广东省江门市江海区海伦心苑1-3203

【导语】在2015年,为了更好的帮助中小企业理解并实践21CFR820.30章节中关于设计控制(DesignControl)的规定,FDA发布了关于设计控制的培训。本文梳理了其中的主要内容,针对FDA对于设计控制的定义、要求等相关内容做出了解析。
什么是设计控制?FDA 给出的定义是:
A set/framework of quality practices andprocedures incorporated into the design and development process.
Control the design process – Premarket and Post-market - to assure that device specificationsmeet user needs and intended use(s).
They set medical device Quality Systemsapart from Good Manufacturing Practices(GMP).
总体上来看,设计控制应该作为质量体系的子系统。设计控制是产品设计研发过程中的一系列质量控制措施,设计控制的目的是通过控制研发过程来确保产品能够持续满足预期用途,同时,设计控制也延续到产品上市生产的过程中。
在FDA的规定中,设计控制适用于所有的II类,III类,以及以下6种I类医疗器械:
Devices automated with computer softwareTracheobronchial suction catheters Surgeon´s gloves Protective restraintsManual radionuclide applicator system Radionuclide teletherapy source
设计控制应贯穿于产品设计开发和生产的每一个阶段。例如在可行性研究/原型机阶段,产品研发阶段,临床试验开始之前,临床试验中及生产开始后产品变更的阶段。
风险管理和分析是设计控制中的重要组成部分以及核心要求。风险管理是对产品质量风险进行分析,评估,并且控制和减轻的系统过程,这一过程贯穿产品整个生命周期,包括若产品发生变更后的风险评估和控制。制造商应该明确产品相关的危害并将相关风险最小化,对相关风险进行判定及评价,控制这些风险,并对控制措施的有效性进行监测。由于在产品开发和使用过程中人为因素影响无处不在,将人为因素和产品风险控制纳入设计控制的考量显得非常重要。【EN ISO要求也包含标识,用户对说明书的理解。】
FDA对于设计控制的具体规定在21CFR 820.30这一章节中。下面对于这个章节的条款进行逐一解析:
1. 21CFR 820.30(a) General --建立并且保持医疗器械设计流程。
•Establishprocedures to control device design •Maintainprocedures to control device design
Establish 和maintain procedures 是2个很重要的概念。Establish指的是建立,形成文件,并且实施。Maintain指的是评审,批准和更新。
2. 21CFR 820.30(b) Design andDevelopment Planning --设计开发计划
每个制造商应建立并保留计划,该计划描述各种设计研发活动,明确执行责任以及影响到设计的活动。其次,这份设计研发计划需要随着项目的进展或变更不断更新。
3. 21CFR 820.30(c) Design Input -- 设计输入
设计输入是指被用作设计基础的器械的物理和性能要求。设计输入部分要求每个生产商都必须建立并保留程序以便确保与器械有关的设计要求是合理的,生产商必须对器械的预期用途进行说明,包括用户和病人的需求等等。设计输入必须处理好任何不完整、模糊或者相冲突的规定。
4. 21CFR 820.30(d) Design Output--设计输出
设计输出是指每个设计阶段工作的结果。对设计输入和输出进行充分的一致性评估并且记录下来,这是一个重要的环节。设计输出应包含具体的可接受标准,以确保器械有效发挥其关键功能。同样,设计输出必须记录成文、得到评审和批准。最终完成的设计输出是器械主文档(DMR)的基础,应作为产品规格包含在上市文件递交中。最终完成的设计输出包括器械、器械包装,标签以及器械主记录。
5. 21CFR 820.30(e) Design Review --设计评审
设计评审是针对设计编制成文的、全面的、成系统化的检查。目的如下:
•评价设计要求是否全面
•评价设计是否能够具有符合所有要求能力
•识别任何可能出现的问题
设计评审应当贯穿于各个适当的阶段并需要将评审结果记录到DHF(设计历史文件)中。设计评审的参与者包括如下人员:
•与接受评审的设计阶段有关的所有部门代表
•与接受评审的设计阶段没有直接责任关系的个人或小组
6. 21CFR 820.30(f) Design Verification--设计验证
设计验证用来验证设计输出符合设计输入,通过提供证据来证实,规定的要求已经满足。(i.e., Input = Output)。设计验证是一个“有限”的概念,这是因为设计输入规定的要求是设计开发策划中确定的,既定的要求,那么需要进行设计验证的对象也是有限的。
7. 21CFR 820.30(g) Design Validation --设计确认
设计确认是提供客观证据证明可以持续的满足用户用途和预期用途。这个定义中包含了持续满足预期用途这一层要求。因为预期的用途不同于具体的设计规定要求,“持续”和“预期”都使得设计确认隐含了“无限”的概念。
可以看出,设计验证阶段提出的问题是“Did I make the product correctly?”而设计确认阶段提出的问题是“Did I make the correct product?”【注:因设计验证和设计确认涉及系统性的话题,在此不过多展开】
8. 21CFR 812.30(h) Design Transfer--设计转化
设计控制规定制造商必须建立并维护流程来确保器械的设计能够正确地转换成成品。完成设计转化环节意味着你的产品结束了研发阶段,正式进入到成品阶段。在经过最终的设计评审之后,要根据设计输出文件编制一份器械主文档(DMR),并确保及时更新设计历史文件(DHF)以应对FDA的审批。
9. 21CFR 820.30(i) Design Changes-- 设计变更
在执行任何设计变更前都应对变更进行识别、记录、验证确认和评审。设计变更是设计控制一个重要的组成部分,发生变更时应该进行评估来确定是否需要为产品递交新的申请,设计变更应该更新DHF,必要时更新DMR。
10. 21CFR 820.30(j) Design History File--DHF(设计历史文件)
DHF是描述成品器械的设计历史的记录汇编。DHF 包含了用来开发器械、附件、主要零件、标识、包装和生产工艺的设计活动。这一节要求每个生产商必须为每个类型的器械建立并保留设计历史文件,而每个器械类型是指根据同一个器械主文档生产的器械。
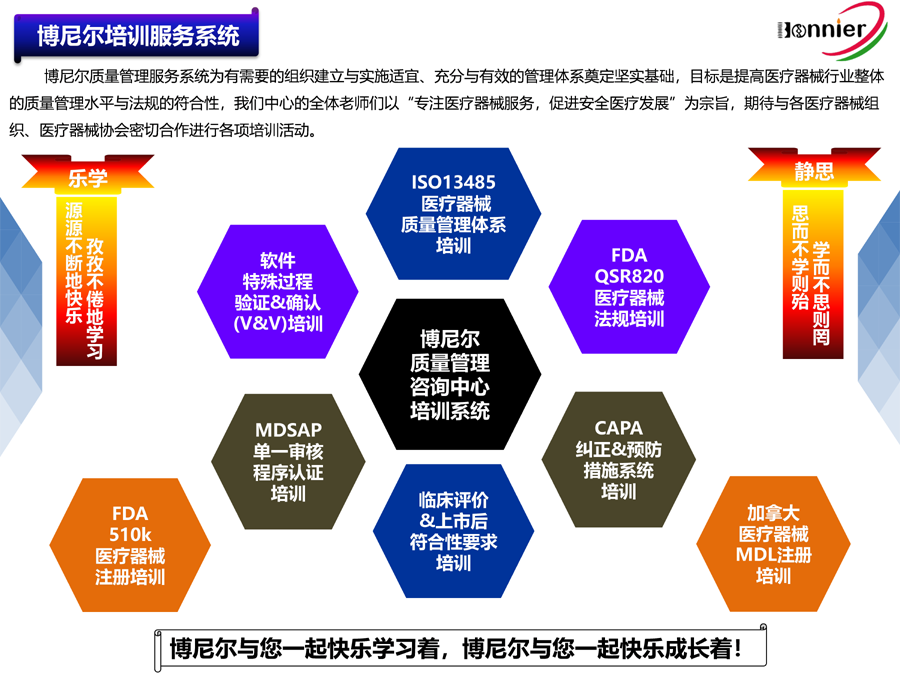
博尼尔的业务范围
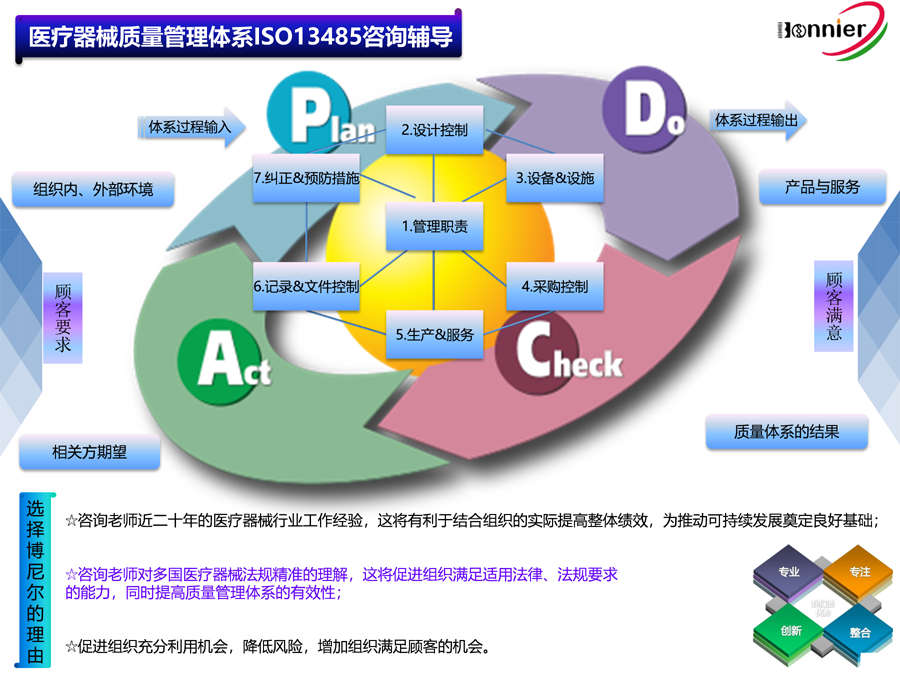
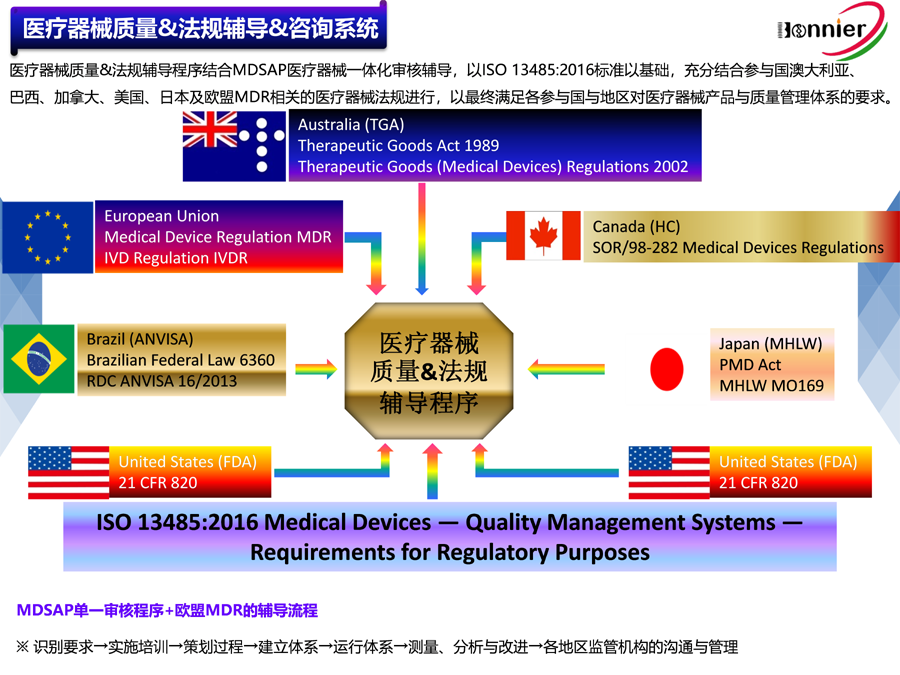
1、医疗器械质量管理体系ISO13485认证咨询与辅导;
2、MDSAP医疗器械单一审核程序认证咨询与辅导;
3、美国FDA QSR820法规体系咨询与FDA验厂服务;
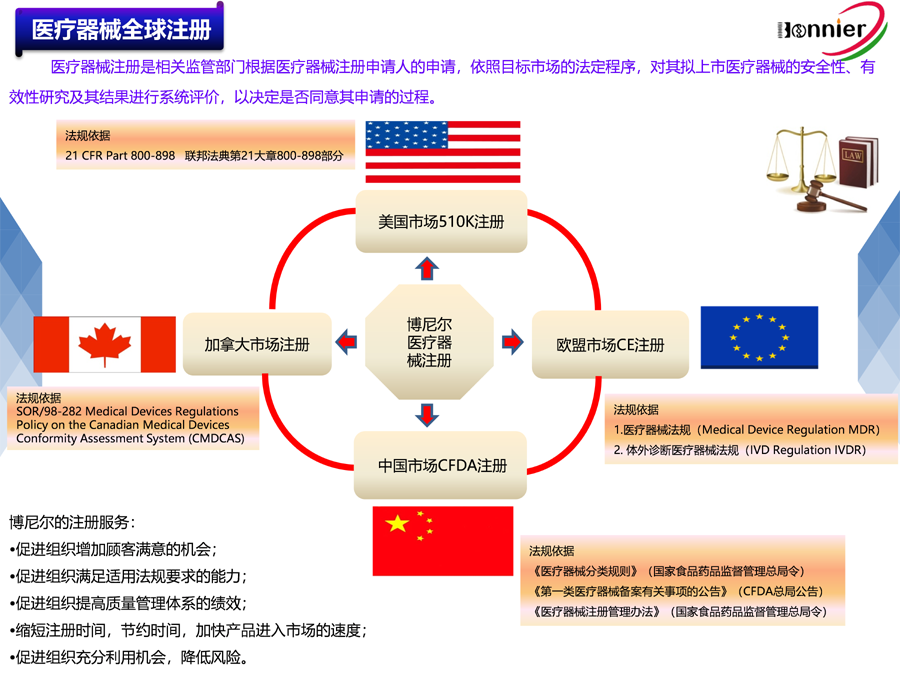
4、全球医疗器械注册代理(美国510k注册、欧盟CE注册、加拿大MDL注册及中国CFDA注册);
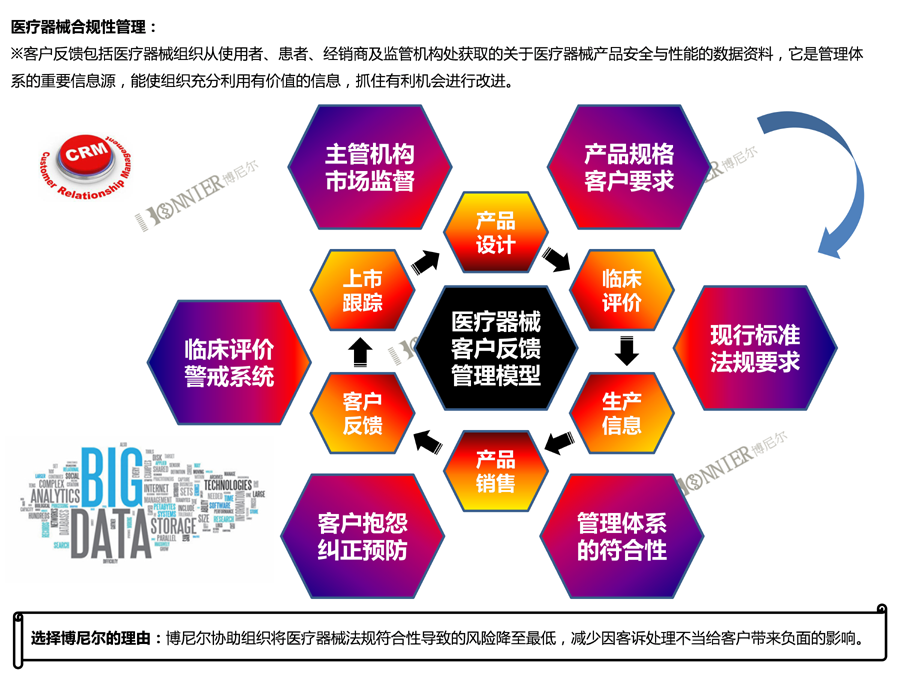
5、医疗器械上市后法规符合性服务(医疗器械不良事件理、医疗器械临床评价及定期安全与性能报告);
6、相关的医疗器械法规与质量工具的培训。