服务热线:
13600366215
13600366215






王老师
13600366215
gary.wang@bonnier.net.cn
Wechat: Cognitive-Level
www.bonnier.net.cn
中国广东省江门市江海区海伦心苑1-3203

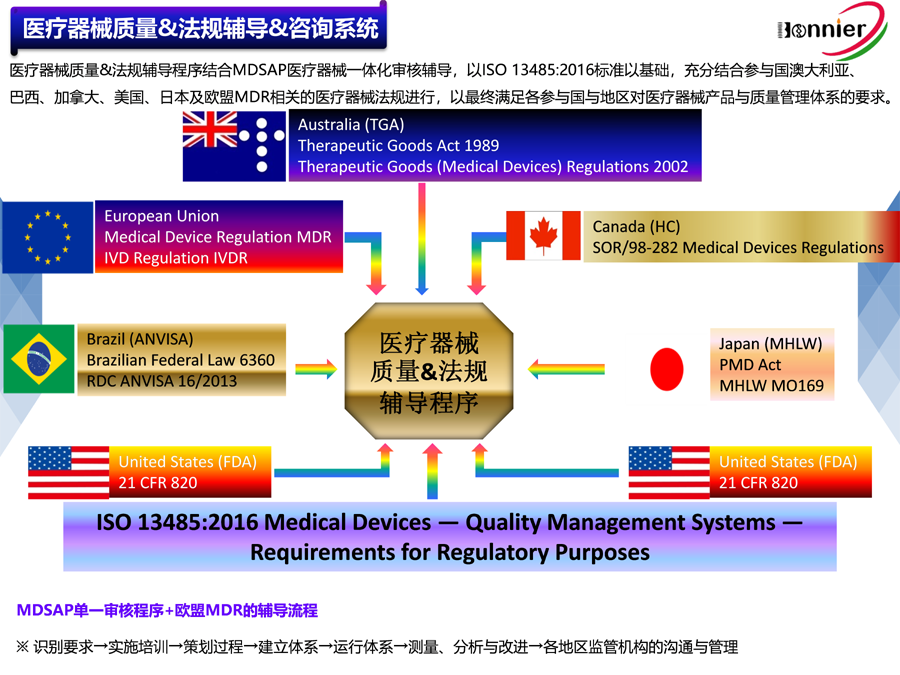
植入性医疗器械因其与人体接触时间长,安全风险高,一直被列为重点监管对象。对于医疗器械而言,生产全过程控制是保障产品安全有效的重要手段,也是国际上普遍采用的管理方式和评价医疗器械质量的基本内容,许多发达国家都把对生产质量体系的审查作为产品能否进入市场的一个重要前提。为了做好医疗器械的监管工作,国家食品药品监督管理总局颁布了《医疗器械生产质量管理规范》,对于风险高、生产工艺复杂的植入性医疗器械,总局还制定了《医疗器械生产质量管理规范附录植入性医疗器械》(以下简称附录),以加强对植入性医疗器械的监管。本文对该附录中的部分行业关注内容进行了讨论,供相关人员参考。
1.相关内容介绍
所谓植入医疗器械,一般是指借助手术全部或部分进入人体内或腔道(口)中,或者用于替代人体上皮表面或眼表面,并且在手术过程结束后留在人体内30 日(含)以上或者被人体吸收的医疗器械。植入性医疗器械种类较多,按材料属性可分为:金属材料、高分子材料、陶瓷材料、复合材料、生物衍生材料、可降解生物材料、组织工程及支架材料等。按照产品结构特点,植入性医疗器械可分为有源和无源两大类。无源产品主要包括骨结合及脊柱结合植入物、骨与关节替代物、心血管植入物、神经外科植入物、乳房植入物、非血管支架、眼内人工晶状体、宫内节育器、齿科种植体、人工皮肤、脱细胞生物膜、整形用透明质酸钠等;有源产品主要包括植入式心脏起搏器与除颤器、植入式人工耳蜗、植入式输液泵、植入式神经刺激器、植入式血泵等。先进科学技术的发展,带动了有源和无源植入物以交叉组合方式进入医疗器械领域,如左心辅助装置或全人工心脏等。
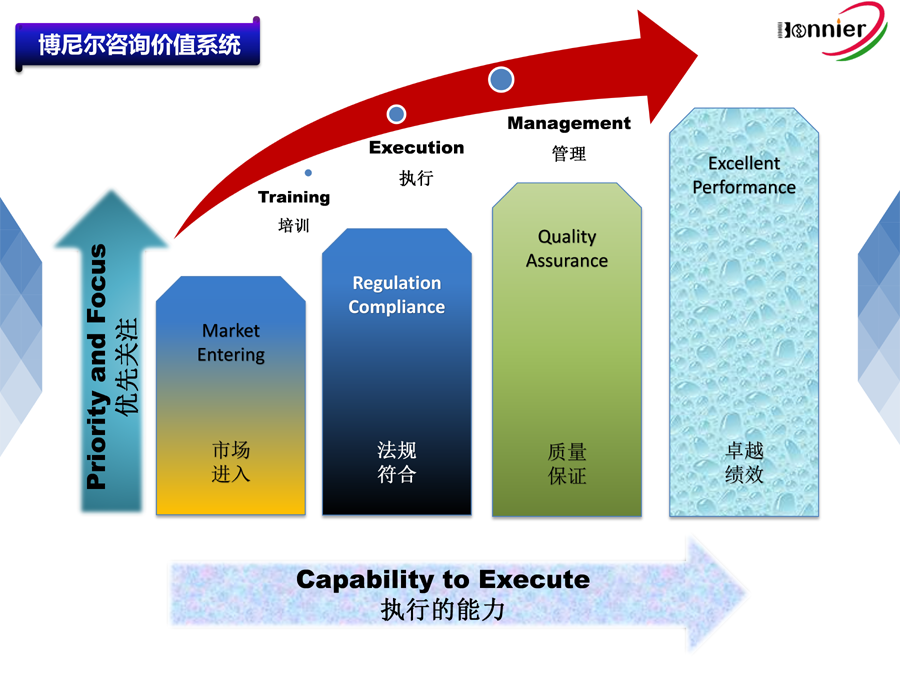
医疗器械生产管理规范是生产企业持续、稳定地提供符合设计要求和质量合格产品的根本保证。在全球医疗器械监管重点从上市前审批向上市后监督、从最终产品质量监督向医疗器械全生命周期控制转移的大趋势下,建立和完善医疗器械生产质量管理体系对于保障医疗器械的安全、有效具有特别重要的现实意义。
2.关于动物源性植入医疗器械的生物安全性
相对于其他种类,动物源性植入医疗器械具有更好的性能,但从另一个角度来看,它们应用到人体可能会增加病毒传播和免疫原性等方面的安全风险。因此,对于动物源性植入医疗器械,在设计开发过程中,应当对原材料及其他生物活性物质的安全性进行充分验证,以最大程度降低产品风险。对于动物源性植入医疗器械而言,生物安全风险主要涉及病毒和/或传染性因子感染以及免疫原性风险三个方面。
(1)病毒和/ 或传染性因子感染
动物种类是病毒和/或传染性因子感染的重要影响因素,主要包括定点饲养、定点采购、定点屠杀,以及根据相关规定进行动物防疫、检疫等。常用的病毒和/或传染性因子灭活方法包括:巴斯德消毒法(巴氏消毒法)、干热灭活法、γ 射线辐照灭活法、过氧乙酸-乙醇灭活法、乙醇灭活法等。免疫原性指能够刺激机体形成特异抗体或致敏淋巴细胞的能力,常见的免疫原性物质包括蛋白、核酸、多糖、脂质、α-Gal 抗原和其他小分子物质等。生产工艺是降低产品免疫原性的主要手段,如脱细胞处理、提纯,以及采用其他物理或化学方法,对具有潜在免疫原性的物质进行去除或对其抗原表位进行消除/隐藏。不过需要注意的是,上述处理措施可能会影响材料的使用性能,也可能会增加新的安全风险。对于动物源性医疗器械,附录条款2.4.2 规定,在研制开发过程中应当对相关材料及生物活性物质的生物安全性进行验证并形成文件。因此,生产企业应当制定用于确保动物源性植入医疗器械安全的控制文件,如动物来源控制文件、生产工艺控制文件等。
(2)动物来源控制文件
包括生物的种属(必要时可确定到品系)、地理来源(对于无法确定地理来来源的种属,可提供来源动物生存其间的识别与追溯信息)、年龄、取材部位及取材部位的组织性质的选择、动物及取材组织的健康状况、所执行的检疫标准、必要的性能控制指标等。
(3)生产工艺控制文件
对于清除(或降低)动物源性原材料免疫原性工艺过程的描述、质量控制指标与验证性试验(例如按照GB/T 16886.20/ISO 10993-20《医疗器械生物学评价第20 部分:医疗器械免疫毒理学试验原则和方法》进行的免疫毒理学试验,参照YY/T0606.25《组织工程医疗产品 第25 部分:动物源性生物材料DNA》进行的残留DNA 检测及α-Gal抗原的检测,或通过其他物理的或化学的试验间接证明产品的免疫原性可得到有效控制)数据或相关资料。
3.关于动物源性供体风险分析管理资料
风险分析是风险管理的基础。对于动物源性植入医疗器械而言,供体的风险无疑是其中非常重要的影响因素,因此在附录条款2.5.4 中,专门对动物源性供体进行了规定,提出生产企业应当对用于医疗器械生产的动物源性供体进行风险分析和管理。按上述条款要求,生产企业应考虑权衡产品的安全风险及与其他替代品比较的预期医疗受益,对动物材料(包括动物种属和组织的选择)的使用进行论证。风险分析应该考虑到以下因素:与医疗器械安全性有关的定性与定量表征的判定;危害和危害处境的判定;在医疗器械的生产中使用有生命的动物组织或者衍生物的具体情况等。 除此之外,还要对动物源性植入医疗器械的典型危险进行相关的风险分析,包括细菌、霉菌或酵母菌污染;病毒污染;传播性海绵状脑病因子污染;不希望的热原、免疫学或毒理学反应方面的材料性反应等,相同的原则也适用于寄生虫或其他未分类的病原体。
YY/T0316/ISO14971《医疗器械 风险管理对医疗器械的应用》以及YY/T 0771.1《动物源性医疗器械 第1 部分 风险管理应用》规定了与该类器械相关的危害与危害处境的判定、估计和评价结果风险、控制这些风险以及监视控制有效性的程序,生产企业可通过风险管理文档验证这些要求的符合性。
对于动物源性材料的质量管理,适当吸取国外经验,逐步把动物饲养作为质量体系的一部分是今后的发展趋势,如果动物源性材料的供方没有建立适当的质量保证体系,或所建立的食品级的质量保证体系不能满足动物源性材料有关标准的要求,生产企业应对供方实行质量体系审核,以保证这些材料满足要求。
4.关于植入医疗器械的可追溯性
可追溯性是一种能力,可还原产品生产全过程和应用历史轨迹,发生场所及销售渠道等。植入性医疗器械需要有这样的能力,一旦发生不良事件,可迅速用于产品召回、分析原因和进一步改进等。另外,对于植入性医疗器械而言,上市前的实验室研究数据、动物试验和小样本的临床数据并不能完全证明产品远期的安全性和有效性,严谨的长期临床随访是其中一个非常重要的证据,而可追溯是实现长期临床随访的重要保证。产品的风险不同,可追溯要求的程度也应有区别,例如对于直接接触心血管系统、淋巴系统或脑脊髓液的产品,应至少能够追溯到产品生产所用的原材料、灭菌设备和生产环境等。美国食品药品监督管理局2010年颁布的《医疗器械追溯要求》,就充分体现了产品之间可追溯要求的差异性。该指南文件对追溯要求的法律法规、需要进行追溯的医疗器械种类,生产、销售、使用等各个环节的部门职责等内容进行了详细的规定。
附录对不同环节的可追溯性也提出了相应的要求。附录条款2.5.5 明确了动物源性产品供体的可追溯性,包括供体单位的资质证明、执行的检疫防疫标准等。附录条款2.6.11规定了生产的可追溯性,包括原材料、生产设备、操作人员和生产环境等。附录条款2.6.12还特别强调了确保产品可追溯的标志,如生产企业名称、批号等。附录条款2.8.1 对产品的销售也提出了要求,代理商或经销商应保存分销记录,企业应保存货运包装收件人的名字和地址的记录等。需要注意的是,对于可追溯性,在不同的环节,附录的要求也不尽相同,不要求每个环节都面面俱到,但要确保发生不良事件时,产品能实现可追溯。
5.几点思考
(1)植入性医疗器械结构组成复杂多样,涉及学科范围广,生产过程和生产工艺差异较大,质量体系的结构、形式和过程不一,因此很难建立一个统一的、详细具体的、要求明确的生产质量管理规范,目前还是以原则性的要求为准。例如附录条款2.4.3,原则上,对于加工工艺过程中使用的各种助剂及残留单体、小分子残留物等,如果后期的加工不能有效去除,都应该进行风险分析,进行毒理学判定,对有毒性的污染物要确定其限量,经检验合格后方可放行。考虑到对于不同的产品,上述物质的风险不尽相同,所以附录仅仅要求对助剂的使用及对杂质的控制情况进行验证,并形成文件,至于如何控制杂质、怎样验证、验证的主要内容、形成文件的类型等,并没有明确要求,生产厂家可依据产品风险制定相应的要求。在确保上述原则的基础上,为加强规范的可操作性,相关部门可参照美国食品药品监督管理局的做法,制定质量体系检查指南,明确检查的路径和检查的方法及步骤,正视产品的差异性,注重系统评估,尽量避免机械式核对条款。
(2)可追溯是个体系工程,而不仅仅是个制度,至少要注册、生产、使用和销售四个环节的无缝衔接,才能实现信息的有效传递和沟通。体系的建立首先需要法规的支持,除注册和生产之外,至少在销售和使用环节,如经销商和医疗机构等,也应建立相应的规章制度,明确其在体系中责任和义务,以确保及时传递正确的产品信息。其次是上市前和上市后产品信息的一致性,譬如上市后的产品,通过UDI(Unique Device Identification,医疗器械唯一标识)、批号或者序列号,可以追溯到生产环节,但是无法追溯到上市前环节,这也是其他国家同样面临的监管难题。目前国际医疗器械监管机构论坛(International Medical Device Regulator Forum,IMDRF)组织已经启动了通用数据元素(Common Data Element,CDE)项目,其主要目的是通过建立统一规范的器械识别信息,确保在医疗器械整个生命周期中信息传递的连续性和有效性,避免监管链条中的“信息孤岛”现象,该项目的研究成果可为我国可追溯体系的构建及与国际接轨提供借鉴参考。
(3)附录条款2.1.1 中,对于从业人员有特殊要求,譬如具有相关的专业背景和实践经验等,也提出了专业培训的要求。对于高风险的植入性医疗器械,考虑到专业性和风险可控性,相关部门可参照药品生产管理规范,设立关键人员制度,如生产管理负责人、质量负责人等,其主要职能是确保产品按照批准的工艺规程生产、储存,确保各项操作规程及其他必要的验证工作得到严格执行,确保产品质量符合经注册批准的要求。
(4)关于产品出厂检验,附录条款2.7.5 提出了产品出厂检验的要求。植入性医疗器械的出厂检验是指通过产品检验的方式,确认产品符合经注册批准的产品技术要求,并能够进行最终产品合格放行的检验。出厂检验并不一定是在成品上进行的,可包括原材料、半成品和成品的检验。出厂检验中的检验项目、检验方法应符合强制性标准及产品技术要求的相关内容,如果出厂检验是在原材料、半成品上进行的,检验方法与标准(或技术要求)规定的方法若有差异,应进行方法学验证,确认其检验结果的一致性。出厂检验原则上不应委托检验,若个别出厂检验项目不能自行检验,需要委托检验的,被委托的检验机构应具有一定的资质。
(5)关于产品留样,附录条款2.7.6 对产品留样仅提出了原则性的要求。留样的目的是以实物备查的方式,追溯所生产的产品在有效期内质量是否符合产品技术要求,为植入性医疗器械在流通或使用环节出现质量纠纷、安全隐患、不良事件认定等提供可靠的支持,也为改进工艺、改进包装、确定医疗器械贮存条件和运输条件、确定医疗器械有效期等提供科学依据。生产企业应根据产品历史质量状况和生产工艺的稳定程度,在质量管理体系文件中制定留样管理办法,规定留样目的、数量、考察或检验的项目等。 留样的数量和形式要根据标准(或技术要求)中理化性能和卫生学项目进行全检的用量来确定,留样方式一般以成品留样为主,也可原料、半成品、产品替代物留样,无论以何种检材留样,留样的数量应至少支持一次质量可追溯检测,留样时间应短于产品的有效期。
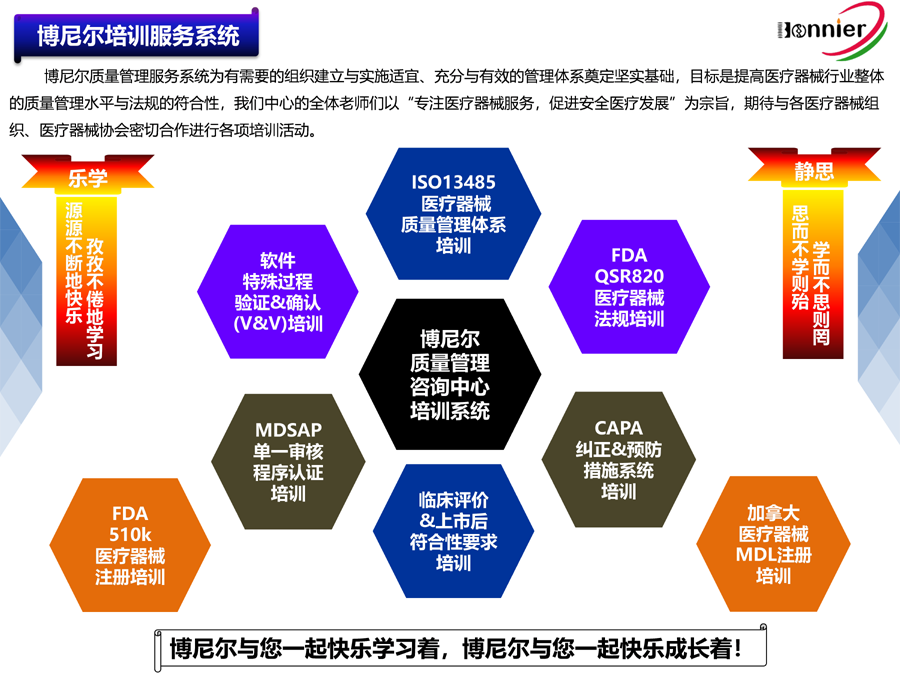
博尼尔专注的业务范围:
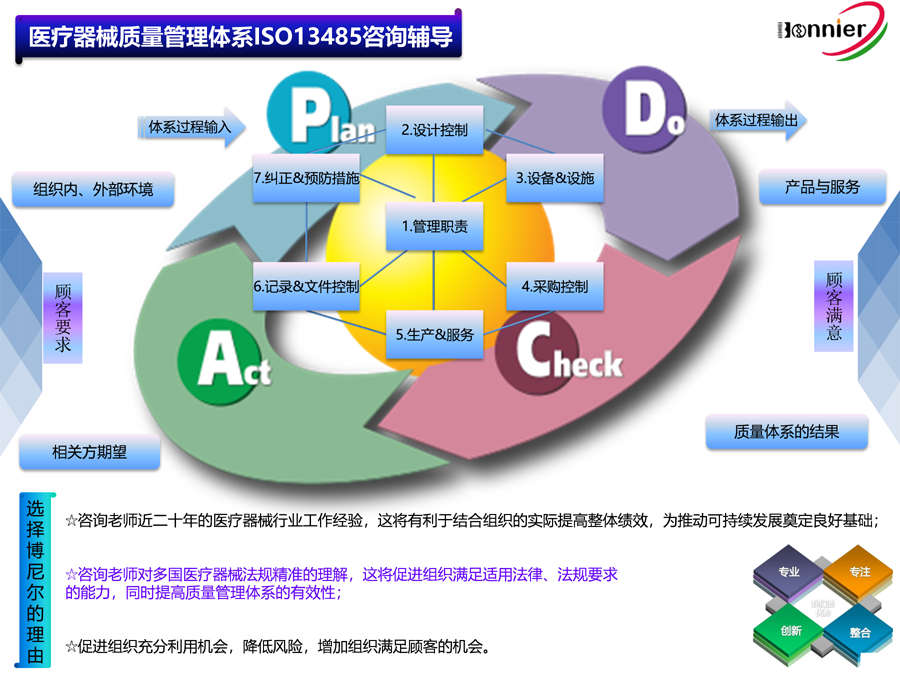
1、ISO13485医疗器械质量管理体系咨询辅导;
2、MD GMP医疗器械良好生产实践咨询辅导;
3、ISO22716化妆品GMP管理体系咨询辅导;
4、MDSAP医疗器械单一审核程序认证咨询辅导;
5、美国FDA QSR820法规体系咨询辅导、协助FDA验厂服务;
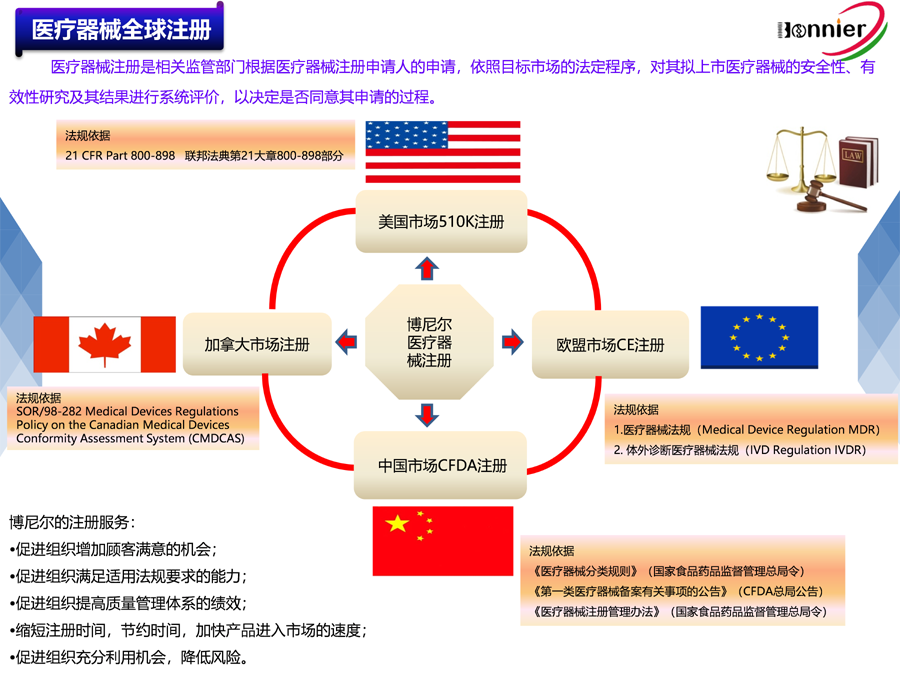
6、医疗器械全球注册服务,包括美国510k注册, 欧盟CE注册, 加拿大MDL注册及中国CFDA注册等;
7、医疗器械上市后法规符合性服务,如医疗器械不良事件管理、医疗器械上市后跟踪服务等;
8、医疗器械法规与质量工具的培训,包括MDSAP法规、CAPA系统、验证&确认、风险管理等;
9、医疗器械中国代理人(MDdelegate in China)服务为海外医疗器械生产企业提供ⅠⅡⅢ类医疗器械在中国的注册及法规符合性服务。